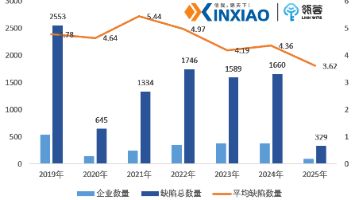
根据2025年FDA(美国食品药品监督管理局)发布的483报告(现场检查缺陷文件)显示,江苏和浙江等省的药企在质量管理中存在较大的风险,缺陷项具体表现在工艺验证、数据完整性、设备管理、质量体系等方面,由此可见,我国药企在质量风险控制中还需持续加强GMP管理。随着我国药品监管标准不断与国际保持同步,企业需要持续提升对于药品质量管理的理论知识和实战经验,提早规避药品过程中可能存在的风险。我们先来看看,历年FDA发布的483报告中常见的缺陷都有哪些?
FDA历年发布的483报告缺陷分析
根据FDA各财年发布的483报告显示,机构人员、设备、生产及工艺控制等方面的相关缺陷项,近几年有占比上升的趋势。2017-2024年483报告中各类别缺陷的出现频次比例
2024财年:指2023年10月1日至2024年9月30日
483报告显示:企业对于制定合理的流程、编写完善的书面程序及遵守操作规程存在较大的挑战统计分析483报告缺陷的具体内容,我们发现,不遵守质量管理流程进行药品生产与风险控制一直是排列在缺陷项的榜首,是企业目前对于药品风险防控的薄弱环节。如何进一步规范人员的行为,提升设备的管理等问题成为药企不可忽视的重点。
2023-2024年483缺陷TOP10对比

为规避以上缺陷,培训对于提高药企的风险管理是非常重要的。尤其是我国GMP管理要求不断与国际相关法规同步的情况下。
GMP无菌药品附录(征求意见稿)发布,提早了解法规变化重点3月17日,国家药监局发布《药品生产质量管理规范(2010年修订)》无菌药品附录(征求意见稿)。
原文链接:https://www.nmpa.gov.cn/xxgk/zhqyj/zhqyjyp/20250317155644123.html
根据《无菌药品附录》(征求意见稿)相关内容显示,修订内容包括污染控制策略的实施、洁净区的动态监管等内容。《无菌药品附录》(征求意见稿)直接借鉴了欧盟附录1的核心条款,系统化引入污染控制策略(CCS)框架,要求企业从厂房设计、人员操作到环境监测全流程识别污染源,并通过风险评估动态优化。条例原文:第八条 制定CCS应当全面考虑,并持续更新和定期回顾,必要时应当对药品质量体系(PQS)进行更新。现有体系如发生变更,应当在变更执行前后评估其对CCS的影响。《无菌药品附录》(征求意见稿)进一步细化了洁净区动态监测的标准,如A级区悬浮粒子有限度豁免≥5.0μm粒子超标。针对以上行业现状,信销科技联合瓴荟智库中心、吉能达一起组织了一场《最新无菌药品监管下的合规策略与先进技术应用》的专题培训,主要内容包括: